DuIvyTools v0.4.8

本文介绍的是v0.4.8及以前的旧版本DIT。
如果你使用的是v0.5.0,请将下文中所有的dit替换成dito执行命令。
Installation
DIT可以通过源码安装(https://github.com/CharlesHahn/DuIvyTools),也可以通过pip安装:
pip install DuIvyTools
目前DIT版本为v0.5.0。
HELP
跟GROMACS类似,DIT专门有个help命令可以输出各个命令的详细信息。以xvg_show举例,可以通过如下命令获取帮助信息:
dit help xvg_show
得到的帮助信息如下:
xvg_show: draw xvg data into line figures.
:examples:
dit xvg_show -f test.xvg
:parameters:
-f, --input
specify the xvg file for input
帮助信息主要分为三部分:第一部分简单介绍命令的功能,第二部分有一两句示例,第三部分则是命令的参数和参数的解释。
当然,为了照顾使用习惯,也可以使用如下方式获得同样的命令帮助信息:
dit xvg_show -h
dit xvg_show --help
dit xvg_show help
除此之外,单独输入dit help会输出DIT支持的所有命令名,方便查找和使用:
$ dit help
******* ** ********** **
/**////** /** ** **/////**/// /**
/** /** ** **/** ** **//** ** /** ****** ****** /** ******
/** /**/** /**/**/** /** //*** /** **////** **////**/** **////
/** /**/** /**/**//** /** /** /**/** /**/** /**/**//*****
/** ** /** /**/** //**** ** /**/** /**/** /**/** /////**
/******* //******/** //** ** /**//****** //****** *** ******
/////// ////// // // // // ////// ////// /// //////
DuIvyTools is a simple analysis and visualization tool for GROMACS result files written by CharlesHahn (https://github.com/CharlesHahn/DuIvyTools).
DuIvyTools provides about 30 commands for visualization and processing of GMX
result files like .xvg or .xpm.
All commands are shown below, type `dit help xvg_show` or `dit xvg_show -h` to show help messages:
XVG:
xvg_show, xvg_compare, xvg_ave, xvg_mvave, xvg2csv, xvg_rama
xvg_show_distribution, xvg_show_stack, xvg_show_scatter
xvg_energy_compute, xvg_combine, xvg_ave_bar, xvg_box
XPM:
xpm_show, xpm2csv, xpm2gpl
NDX:
ndx_show, ndx_rm_dup, ndx_rm, ndx_preserve
ndx_add, ndx_combine, ndx_rename
MDP:
mdp_gen
Others:
find_center, pipi_dist_ang, hbond, mol_map, dccm_ascii
Matplotlib Style:
show_style
You can type `dit help <command>` or `dit <command> -h` for more help messages
about each command, like: `dit help xvg_show` or `dit xvg_show -h`.
And you can also modify the style of figures by adding (only) one mplstyle file to your working directory. DIT will apply it to custom figures. You could
modify mplstyle file based on the template generated by "dit show_style", or
select one from style folder of DuIvyTools github repo
(https://github.com/CharlesHahn/DuIvyTools).
Have a good day !
XVG
XVG模块目前包含了13个命令,命令以xvg开头,都是用于处理xvg格式的文件的。
xvg_show
读文件绘图,默认第0列是X值,第1列及之后的是数据。如果有多列数据,就绘制成子图的样式。
注意,DIT中列和行的计数都是从0开始的!
dit xvg_show -f rmsd.xvg
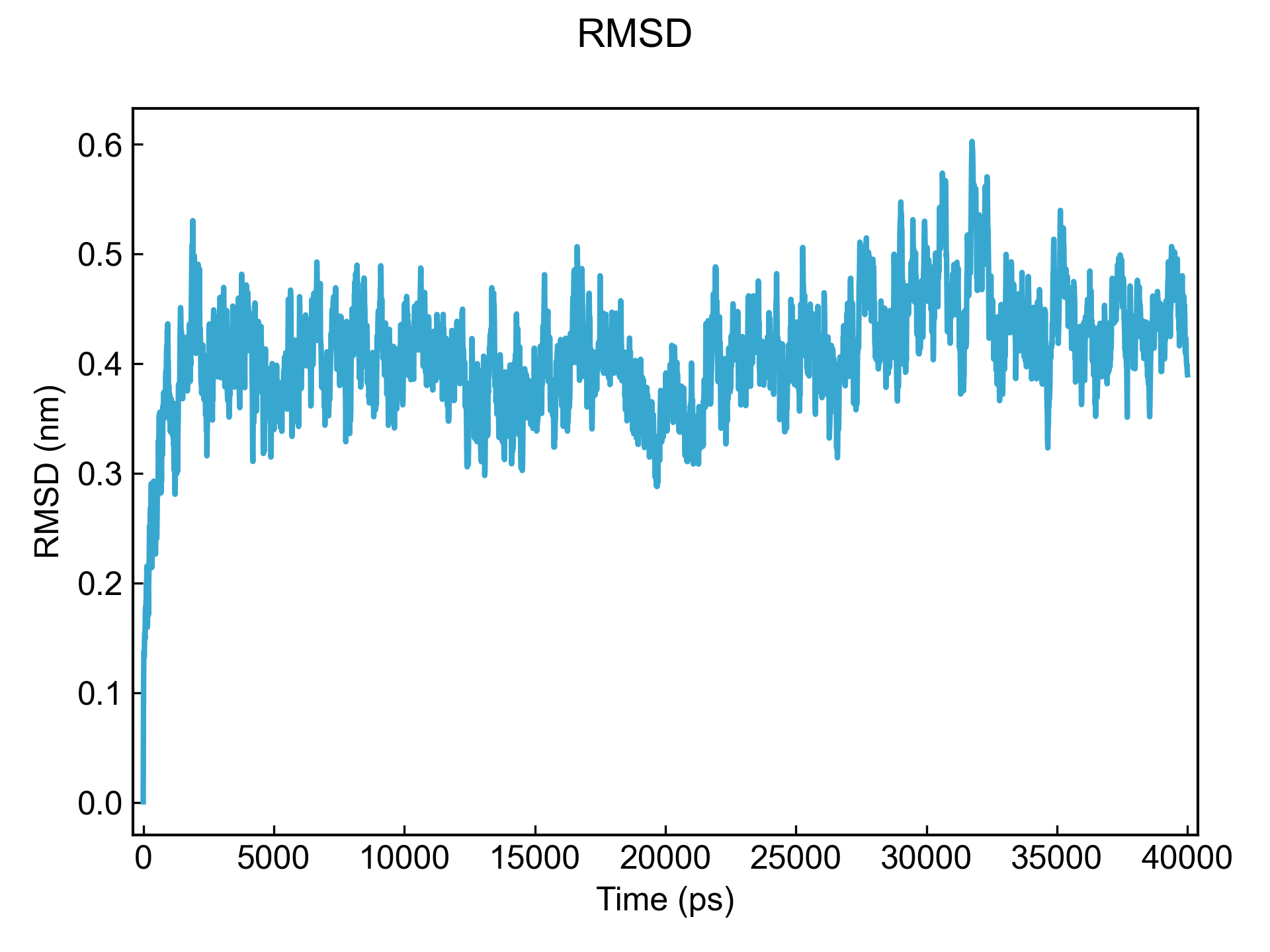
xvg_compare
比起xvg_show,我很推荐使用xvg_compare。即使不需要数据的比较,这个命令的灵活性也更大些。
xvg_compare,提供一个或几个文件,提供一组列索引,即可把索引到的列的数据都绘制出来。
dit xvg_compare -f rmsd.xvg gyrate.xvg -c 1 1,2
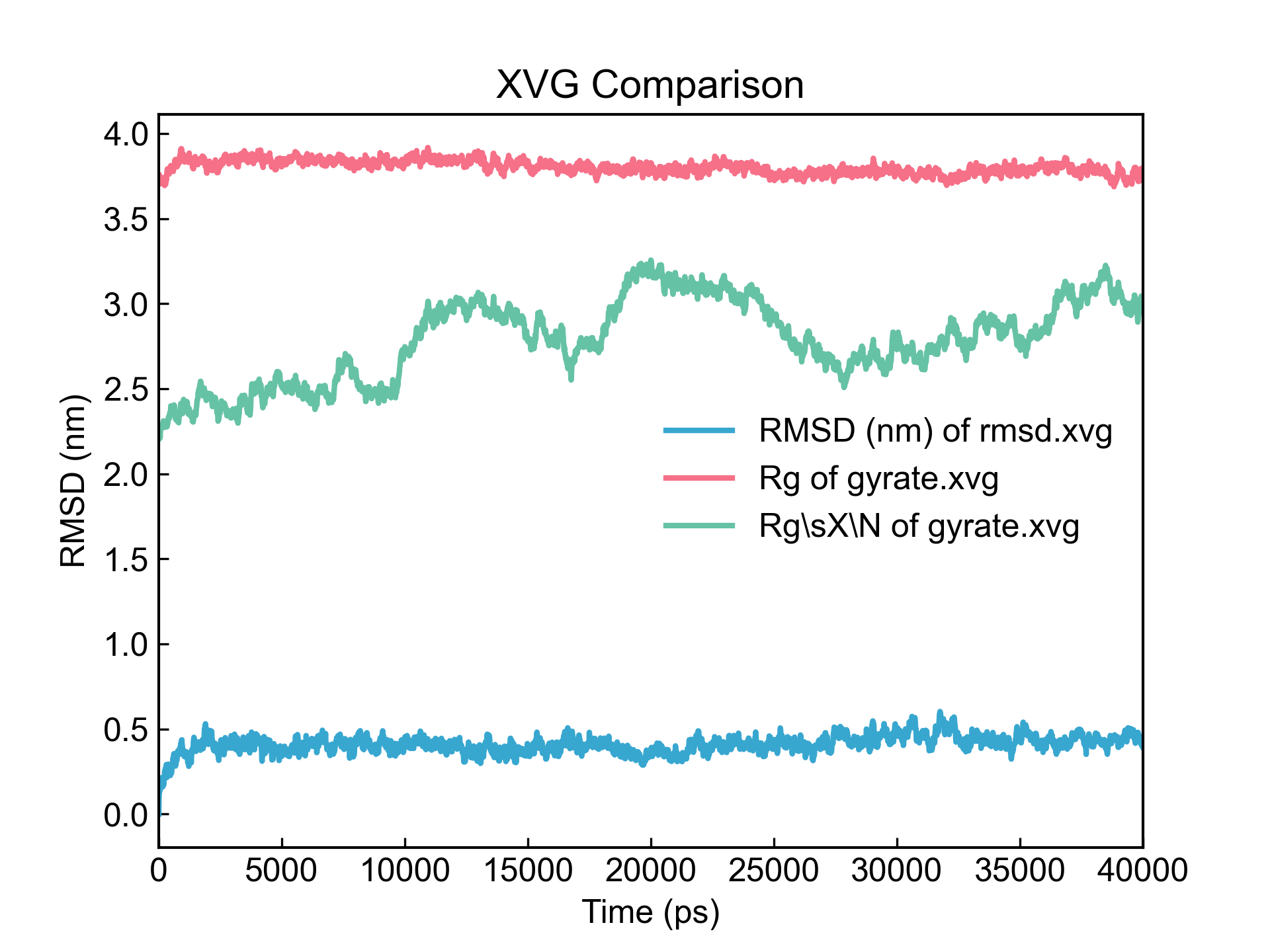
这里的索引可能有些奇怪,为啥是1 1,2。这里实际上是两个索引(一个索引是1,还有一个是索引组1,2)。前面赋给了两个文件,所以这里索引(索引组)的数量得和前面的文件的数量对应,也即是两个索引。索引(索引组)之间用空格隔开。同一个索引组里的索引,如1,2,用逗号隔开。第一个索引1对应第一个输入文件的第一列数据,在这里也就是RMSD;第二个索引1,2对应第二个输入文件,也即Gyrate数据的Rg和RgX这两列数据。
这样的索引的赋值方法,可以使得在每一个输入文件里选择不同的列进行比较。
需要注意的是,输入文件的第0列不一定要完全相同。这里的折线图的绘制,默认就是以对应文件的第0列为X值的。RMSD的第1列数据是以rmsd.xvg的第0列数据作为X值,Gyrate的第1,2列数据是以gyrate.xvg的第0列作为X值的。
xvg_compare这个命令还有其它的参数。虽然legend、xlabel、ylabel等都可以自动解析和赋值,大多数情况下也还能看,但是也可以通过参数手动赋值。
-l参数可以用于图例的赋值,参数的数量和选择的列的数量要保持一致,且不需要对应文件了。目前还不支持在图例里有空格,空格在这里是区分不同图例的标志。我会在下一个小版本里改进,用一个标识符表示空格。
dit xvg_compare -f rmsd.xvg gyrate.xvg -c 1 1,2 -l RMSD gyrate gyrate_X -x Time(ns) -y (nm) -t hhh
还可以通过-s、-e,也就是start和end来设定每一列数据的使用范围,也即起止行数,比如说-s 10 -e 100就是指只使用每一列数据的第11个到第100个(包含)数据。
这个方法里面还包括了求滑动平均的选项,声明一下-smv就可以了。求滑动平均有两个参数需要解释下:窗口宽度(windowsize)和置信度(confidence),默认是50和0.9,当然也可以自己赋值确定。
dit xvg_compare -f energy.xvg -c 1,3 -l LJ(SR) Coulomb(SR) -smv
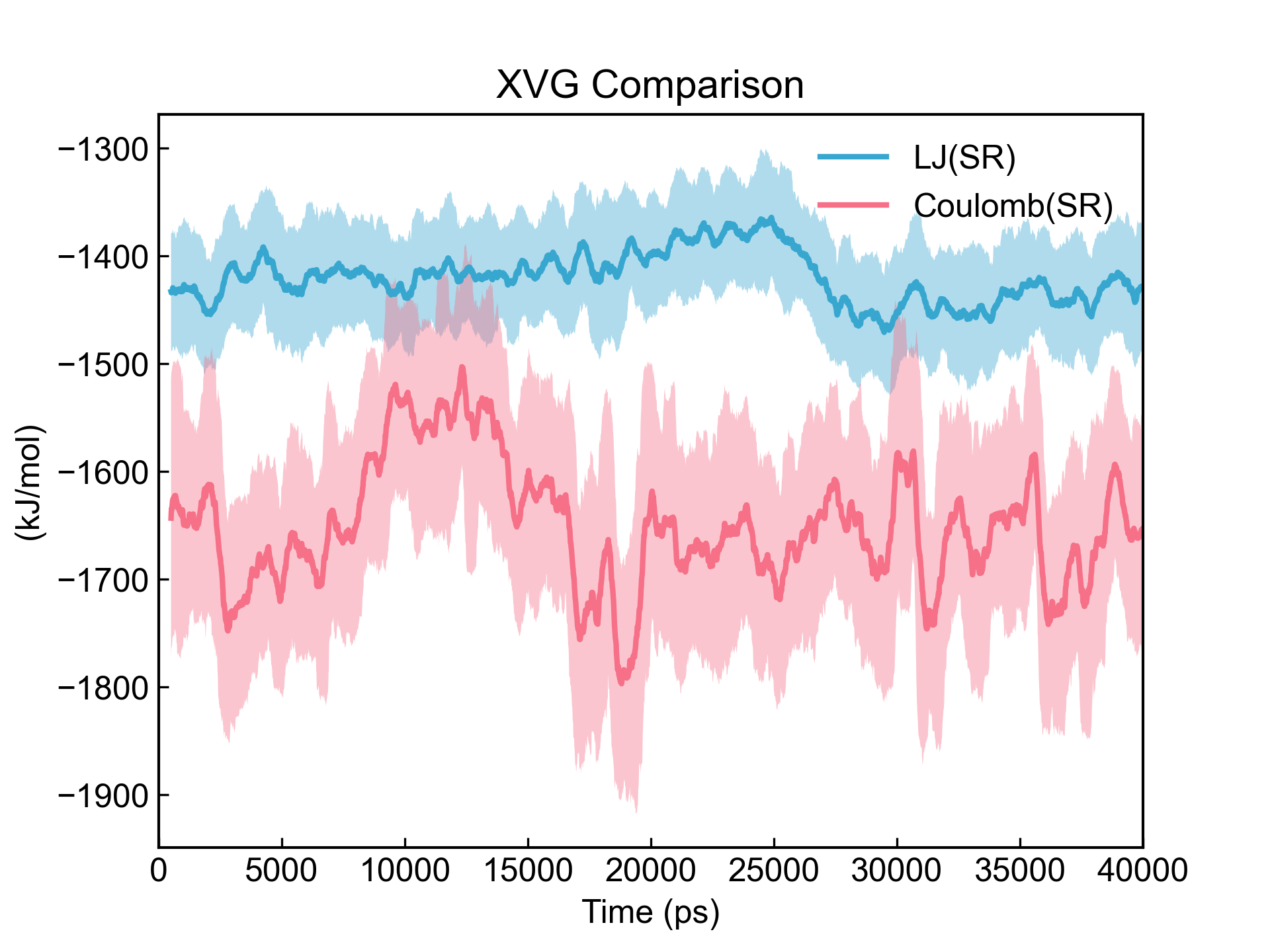
滑动平均的计算有点儿慢现在,后续再优化吧。
xvg_ave
xvg_ave就是用来求每一列数据的平均的,当然你也可以声明起止行数。用来求平衡时期的某些数据的平均值还是挺好用的。
$ dit xvg_ave -f rmsd.xvg -s 2000 -e 4000
Info -> read rmsd.xvg successfully.
Time (ps) RMSD (nm)
ave 29995.0000 0.4281
std 5773.5020 0.0473
需要注意的是,-s声明的起始行是被包括在计算数据中的,而-e声明的行则不被包括。如果你要计算到列的末尾,那就只声明-s就行了。比如说这里的rmsd.xvg一共就4000行,声明了-e 4000之后,实际上只从2000行计算到了3999行,最后一行没有被包括。再次说明,DIT中列和行的计数,都是从0开始的。
xvg_mvave
给定一个xvg文件,求出每一列数据的滑动平均数据,并输出到csv文件。
xvg2csv
把xvg数据文件转化成csv文件,声明输入输出文件就可以了。不再赘述。
xvg_rama
gmx的rama命令是可以得到蛋白质的二面角(phi和psi)数据的,xvg_rama命令就是把这样的数据转换成拉式图。这个命令的大部分内容借鉴自PyRAMA项目。
dit xvg_rama -f rama.xvg
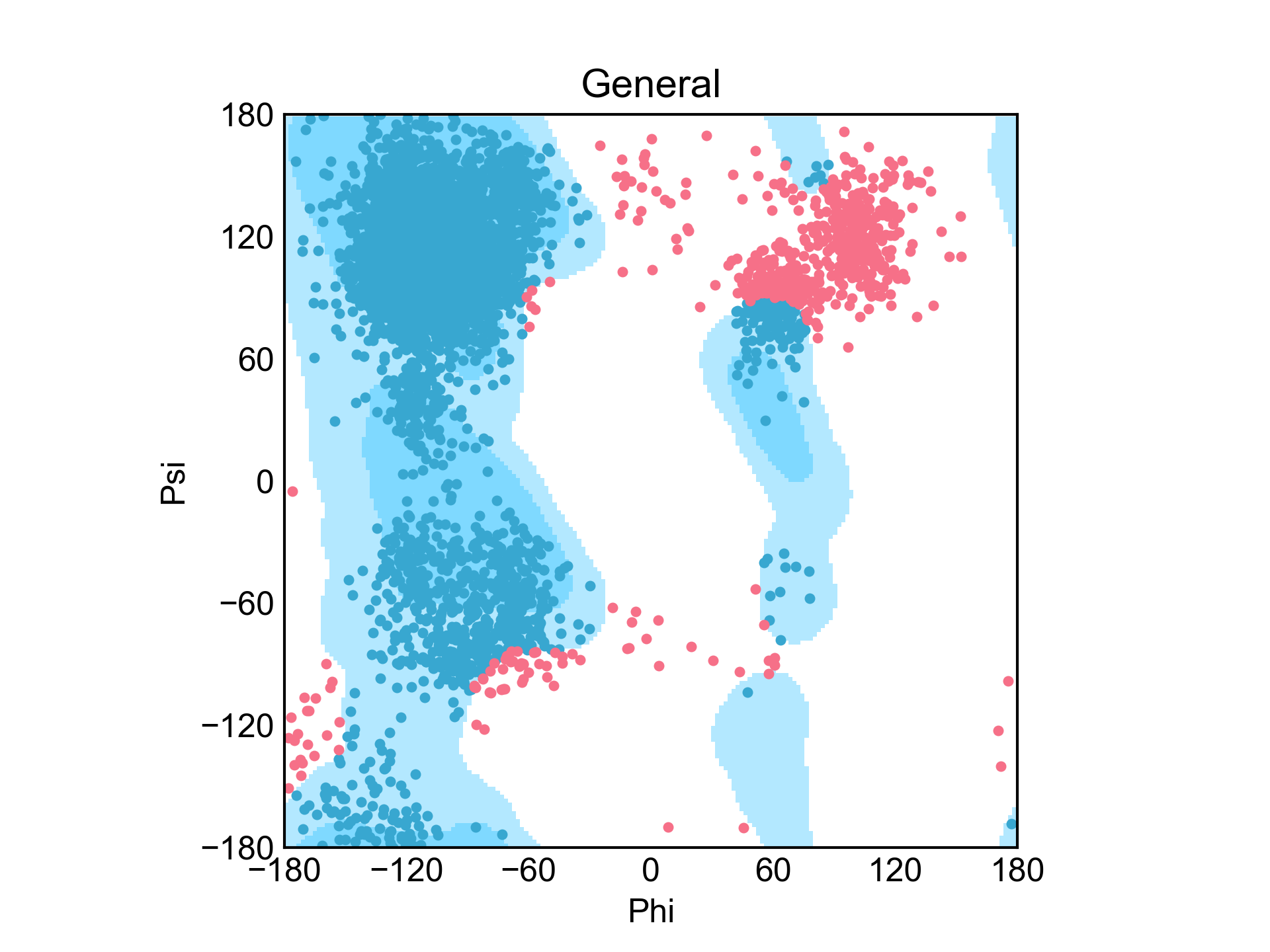
xvg_show_distribution
xvg_show展示数据的变化,xvg_show_distribution展示数据的分布,如果有多列数据的话,也是会绘制成组图。
dit xvg_show_distribution -f gyrate.xvg
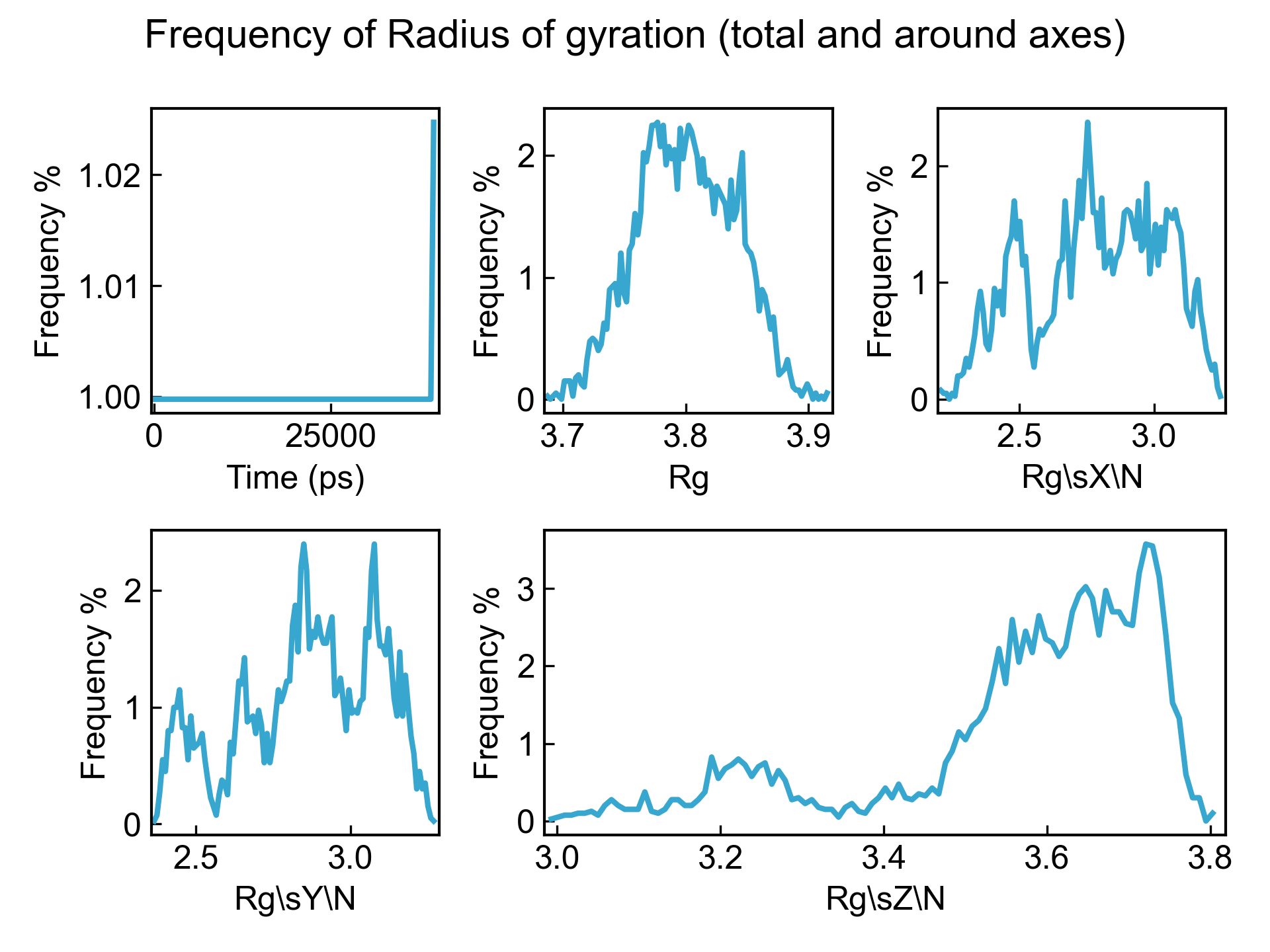
可以通过-bin参数定义要求的分布的格子数,默认是100。最大的数据会归到最大的bin里面,这也就导致了第一个图时间的分布最后有个小的抬头。
xvg_show_stack
xvg_show_stack命令主要是绘制堆积折线图的。有时候需要绘制蛋白质二级结构含量的变化,就可以用这种堆积折线图对do_dssp命令得到的二级结构含量xvg文件进行绘图。
dit xvg_show_stack -f dssp_sc.xvg -c 2 3 4 5 6
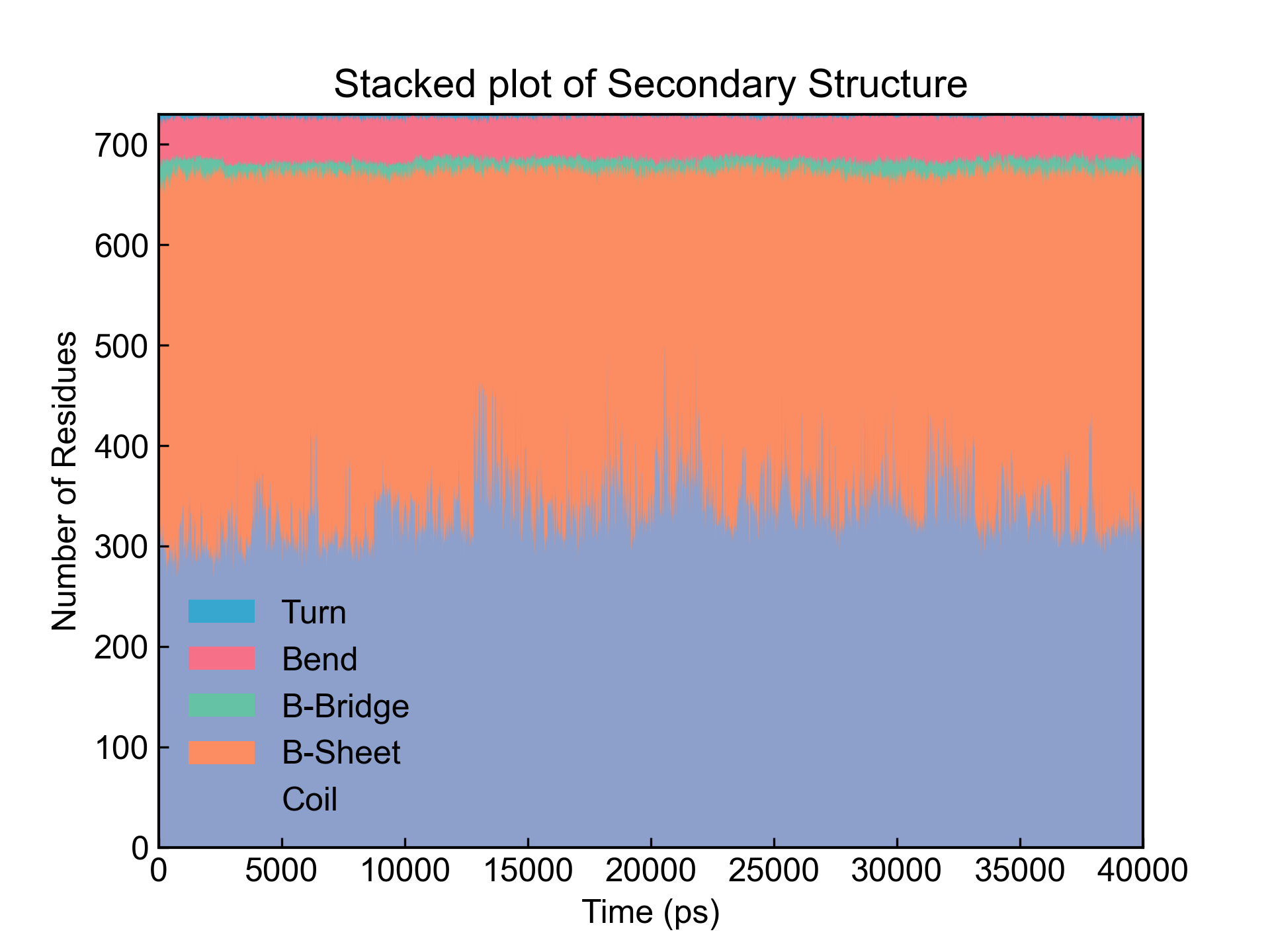
用-c参数指定要堆积的列,程序会自动将选中的列绘制为堆积折线图。
xvg_show_scatter
不消说,xvg_show_scatter是绘制散点图的,通过-xi、-yi指定两列,分别为X值和Y值,然后绘制成散点图。
dit xvg_show_scatter -f rama.xvg -xi 0 -yi 1
xvg_energy_compute
分子间相互作用,如果你用基于相互作用原理的方法计算的话(相互作用能 = 复合物能量 - 分子A能量 - 分子B能量),这个命令可以帮你快速执行这一过程。
输入三个文件,复合物能量文件、分子A能量文件、分子B能量文件;每个文件应包含且只包含五列数据(时间、LJ(SR)、Disper.corr.、Coulomb(SR)、Coul.recip.),顺序也要正确。这个脚本会读入这三个文件,然后执行计算,输出计算结果到xvg文件。
dit xvg_energy_compute -f prolig.xvg pro.xvg lig.xvg -o results.xvg
当然现在我不太建议你用这个方法了。比较推荐你设置能量组,增大cutoff直接rerun,得到的数据似乎还更准确一些。
xvg_combine
xvg_combine和xvg_compare的命令输入方式类似,功能也类似。xvg_compare将你选中的数据绘制出来,xvg_combine则是将你选中的数据重新输出到一个新的xvg文件中。
dit xvg_combine -f f1.xvg f2.xvg -c 1,2 2,3 -o res.xvg
xvg_ave_bar
xvg_ave_bar就可复杂了,光是说起来就得费一番口舌。
我们假设这样一个场景:你模拟了三个不同的配体分别与蛋白的相互作用,每一个体系都进行了三次平行模拟,这样你就一共有9个模拟轨迹,相应的有9个蛋白与配体的氢键数量随时间变化的xvg文件。现在咱们需要把每一个模拟体系的稳定时期的平均氢键数量计算出来,然后做体系之间的比较。
按照通常的做法,我们需要先对每一个xvg文件中稳定时期的氢键数目求个平均值,一共有9个平均值。然后把同样体系的三次平行的平均值再求个平均,以及误差。最后把三个体系的平均值和误差作成柱状图。
没错,xvg_ave_bar大概就是干这个活儿的。
先看示例,再具体解释各个参数的含义。
dit xvg_ave_bar -f f1_1.xvg,f1_2.xvg,f1_3.xvg f2_1.xvg,f2_2.xvg,f2_3.xvg -c 2 3 5 -l A B -y number
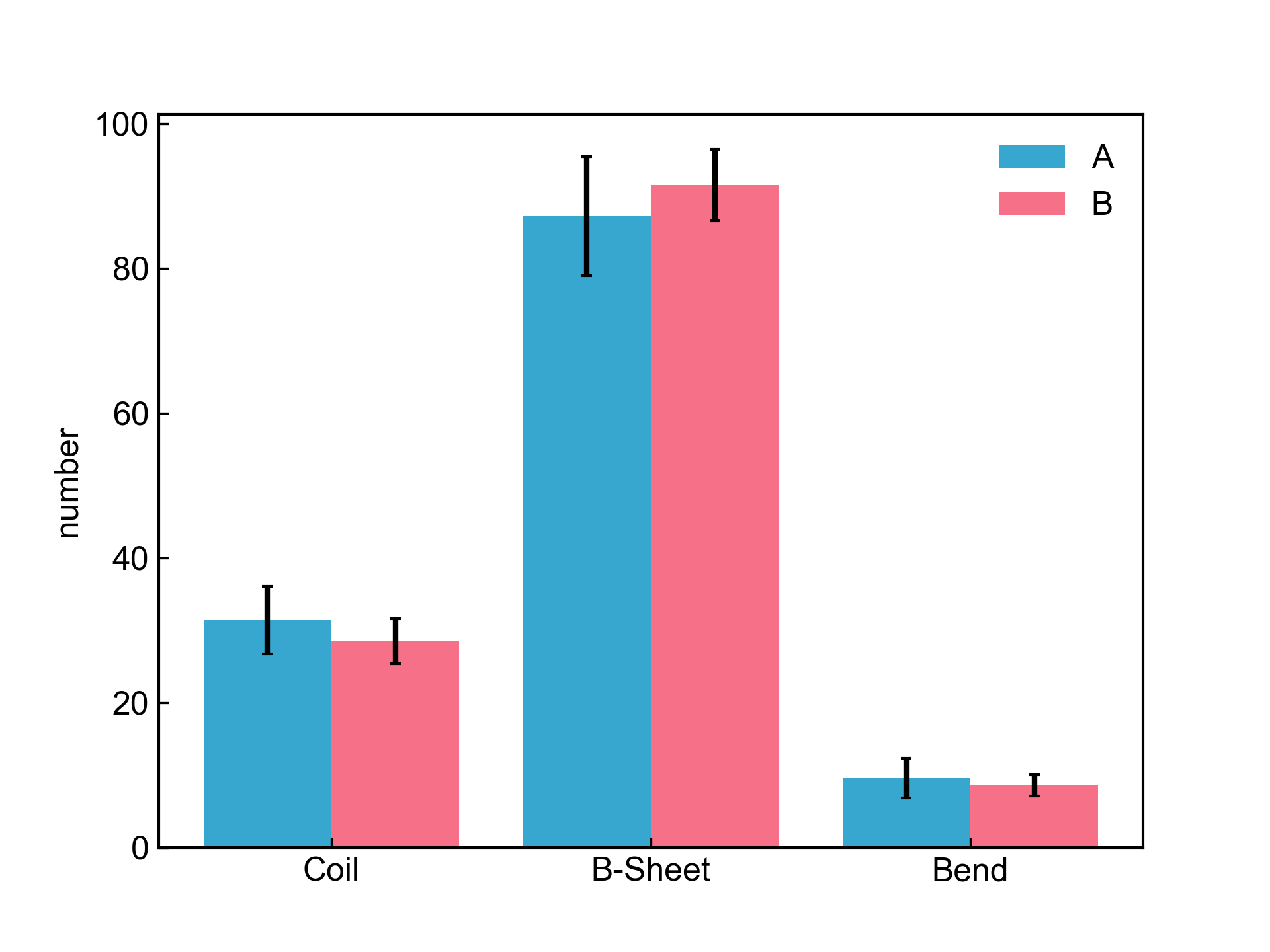
上图绘制的是两个体系(各有三次平行模拟)的蛋白的二级结构含量图。
首先需要解释的是-f参数,这个参数读入若干个文件组,文件组和文件组之间用空格隔开;一个文件组就表示一个体系。每一个文件组内部可以有多个文件,文件名和文件名之间用逗号隔开,这表示一个模拟体系的多次平行模拟。程序会自动对同一个文件组内的多个平行模拟的数据文件求平均和误差,然后绘图进行不同文件组之间的比较。上图有两个体系,A和B,每个体系内有3个平行模拟的数据文件。
-c参数呢就是选择要对比的数据列了,这个参数的值会应用到每一个文件中。比如这里,就是选择了每一个文件的2、3、5列进行绘图,因而最终的图上会有三个X值。
-l参数是用于手动指定图例的,图例的数量要和文件组的数量一致!
-xt参数用于指定在X轴上显示的内容,数量与选择的列数一致。
还可以指定xlabel、ylabel、title、要包括的数据的起止等等。
值得一提的是,有一个-ac参数,指定了这个参数,搭配-o参数指定输出文件名,可以把计算得到的平均值和误差等数据输出到文件中,默认是输出到屏幕上的。
xvg_box
xvg_box会对数据绘制箱型图,比较数据分布情况。
dit xvg_box -f f1.xvg f2.xvg -c 1 2 3 -xt Rg Rg_x Rg_y
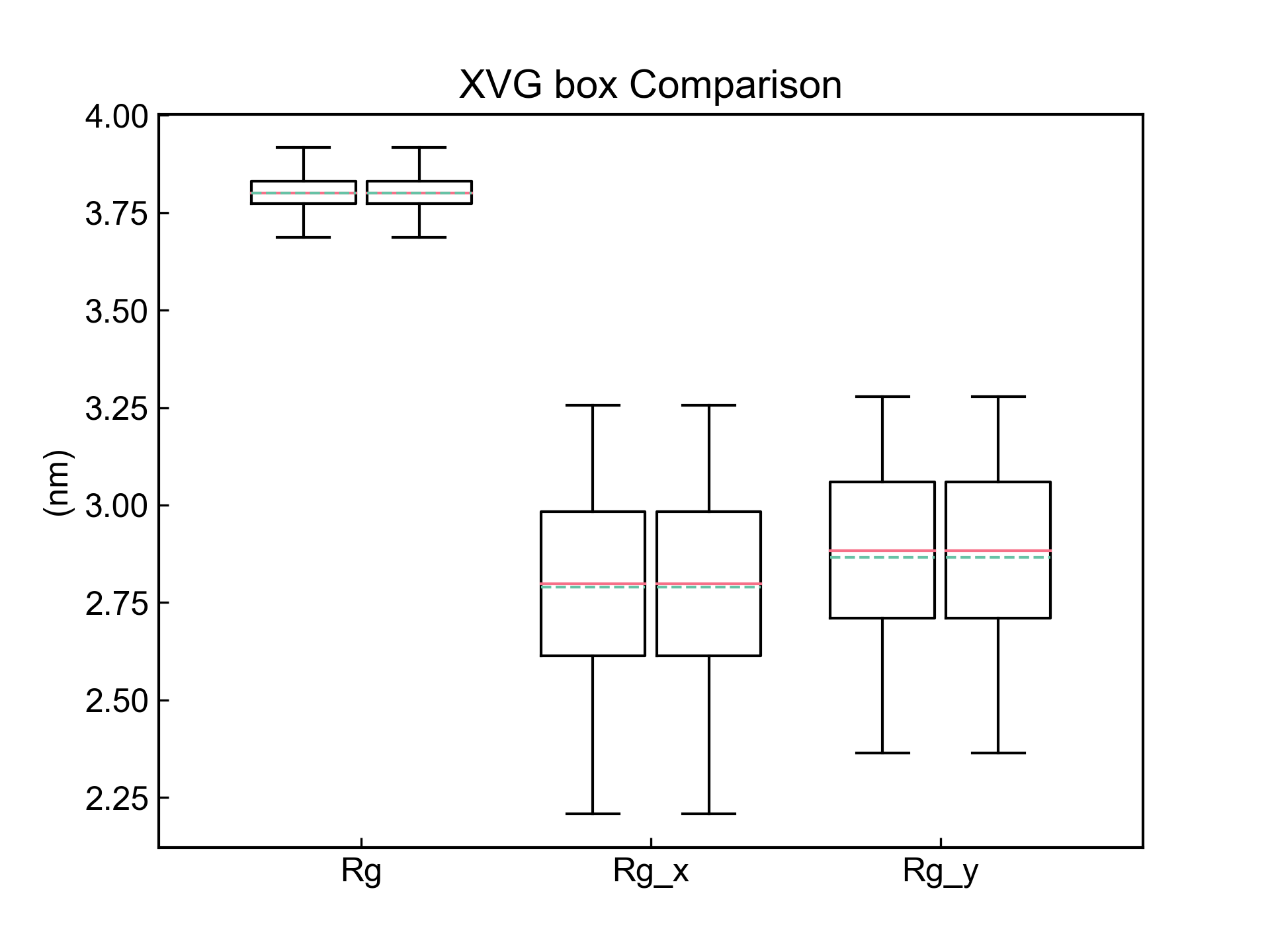
-f参数指定几个文件,然后-c选定几个列(选定的列索引会应用到所有指定的文件中),就可以绘制出箱型图。
需要注意的是,这里需要保证每个xvg数据文件里面的相应的列表达的是同一个意思,比如这里的文件的第1列都是Rg,之后两个列都是Rg_x和Rg_y。程序不会检查有没有对应上。
-xt参数用于声明X轴上显示的值,数量与-c一致。还可以指定xlabel等等。
我想这个功能估摸着是用不到的,写得草率。
XPM
XPM模块包含3个命令,都是处理XPM文件的;命令以xpm打头。
xpm_show
xpm_show用于可视化xpm文件,-ip指定要不要插值,-pcm指定要不要使用pcolormesh函数绘图,-3d指定要不要绘制3d图,-o参数可以用于保存图片文件,-ns参数可以指定不显示图片。
dit xpm_show -f test.xpm -ip
dit xpm_show -f test.xpm -3d
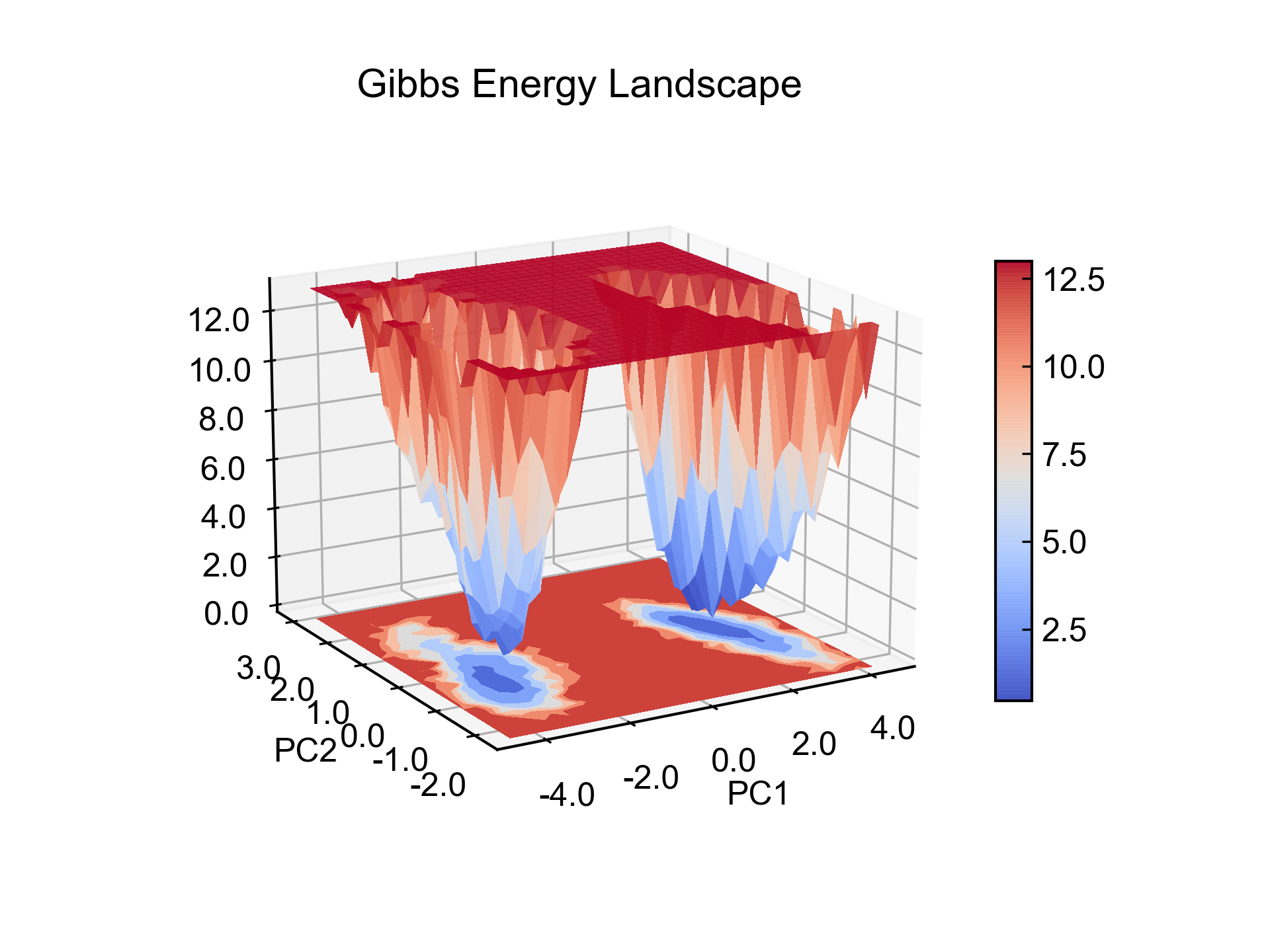
基本上所有GMX生成的XPM图都可以用xpm_show可视化,包括dssp的图和hbmap的图。
xpm2csv
xpm2csv可以将xpm文件转化为csv文件,也即三列数据(x, y, v),横纵坐标以及对应的像素点的值。
dit xpm2csv -f test.xpm -o test.csv
xpm2gpl
Gnuplot是一个很好的绘图软件,它可以通过脚本进行绘图。xpm2gpl命令则是把xpm文件转化为gnuplot的绘图脚本gpl文件(不一定后缀得是gpl,只是这里为了方便定义为了gpl)。用Gnuplot绘制出来的蛋白质二级结构变化图是很好看的。
dit xpm2gpl -f test.xpm -o test.gpl
xpm2dat
将xpm文件转化为M*N的dat文件。目前只支持Continuous数据类型的xpm文件。输出文件的第一行为注释,依次为数据第一行的标题,数据第二行的标题,以及数据M*N矩阵的标题。 数据的第一行为xaxis数据,也即xpm图片的x轴数据。 数据的第二行为yaxis数据,也即xpm图片的y轴数据,顺序为从下到上。 后面的数据皆为M*N的数据矩阵的数据。数据的顺序和xpm文件中的顺序一致,矩阵中上面的数据对应着yaxis中后面的数据。
dit xpm2dat -f test.xpm -o test.dat
NDX
NDX模块共7个命令,以ndx开头,用于处理ndx文件。
我比较推荐你使用gmx的make_ndx命令处理ndx文件,功能更丰富,也更好用。
ndx_show
ndx_show命令会输出ndx文件中所有的索引组的名字。
dit ndx_show -f test.ndx
ndx_rm_dup
ndx_rm_dup命令会删除ndx文件中所有重复的索引组。这里的重复的索引组是指不仅名字一样,索引也一样的组。
dit ndx_rm_dup -f test.ndx -o res.ndx
ndx_rm
ndx_rm用于移除索引文件中的某些索引组,你可以通过-gl参数指定几个索引组的名字,ndx_rm会把这些指定的索引组移除,保留其它的索引组。还可以通过-int参数激活互动模式,可以在互动模式里决定是否删除某一个索引组。
dit ndx_rm -f test.ndx -o res.ndx -gl Protein Ligand
dit ndx_rm -f test.ndx -o res.ndx -int
ndx_preserve
ndx_preserve和ndx_rm基本类似,只是被选中的索引组不再被删除,而是被保留;未被选中的索引组才会被删除。同样也有互动模式。
ndx_add
ndx_add可以给索引文件增加一个索引组,需要你通过-gn执行索引组的名字,-s、-e、-t三个参数指定索引数字的起始、结束和步长。如果你熟悉python的range函数的话,应该很好理解这三个参数的作用。
dit ndx_add -f test.ndx -o res.ndx -gn hhh -s 10 -e 100 -t 2
上述的命令添加一个叫hhh的索引组,数字是从10开始一直到100(不包括)的偶数。-t也可以缺省,默认步长是1。
有的时候(比如做dPCA)需要自己手写一个索引组,因而ndx_add或许还有点儿用途。
ndx_combine
ndx_combine可以组合几个索引组到一个新的索引组,需要指定-gn和-gl两个参数。
ndx_rename
ndx_rename主要是给索引组改名字的,可以-int互动模式更改,也可以指定旧名字-on和新名字-nn来更改名字。
MDP
MDP模块就一个命令了。
mdp_gen
mdp_gen命令可以帮助你在当前工作目录生成一个模板mdp文件。有个-a参数,用于指定mdp文件的用途,根据用途不同,会生成不同的文件内容。目前-a可以接受如下几个参数中的一个:ions、em、nvt、npt、md、blank。比如说:
dit mdp_gen -o npt.mdp -a npt
上述命令会在当前目录生成一个可用于npt的mdp模板文件。
需要说明的是,这里生成的mdp模板文件不一定适合你的体系,请生成之后一定打开自行设置和调整相关的参数。这个命令只是帮你免去把mdp文件复制来复制去的工夫。
这里的这几个mdp模板文件实际上都存储在DuIvyTools这个第三方包目录的data文件夹下,你也可以根据你的需要直接修改这里的文件。
这部分功能后续也将继续优化,我希望能集合大家用于不同模拟方向的mdp文件,凑在一起,方便大家使用。
Other Modules
show_style
dit show_style
show_style命令是v0.4.7新加入到DIT,执行这个命令会在当前工作目录生成一份DIT默认的matplotlib style sheet,也即绘图的样式控制文件。用户可以根据自己需要更改。
matplotlib style相关的参数可以参考 https://matplotlib.org/stable/tutorials/introductory/customizing.html#the-matplotlibrc-file
当然,用户也可以自己找一个样式控制文件(.mplstyle)置于当前工作目录,DIT会优先读取并绘图。
find_center
find_center: to find the atom which is nearest to center of atom group.
:examples:
dit find_center -f test.gro
dit find_center -f test.gro -n index.ndx
dit find_center -f test.gro -n index.ndx -aa
:parameters:
-f, --input
the gro file which contains one frame of molecule coordinates
-n, --index
the index file which you could select group from
-aa, --AllAtoms
if to find center of one group atoms in all atoms
find_center命令主要用于寻找gro文件中组分的几何中心。
用户可以通过指定索引文件和索引组以寻找特定组的几何中心。如果参数中不包括索引文件,则默认寻找整个gro文件所有原子的几何中心。
-aa参数的意思是是否在全体原子中寻找指定原子组的几何中心。有的时候,距离指定原子组的几何中心的原子不一定出现在该组中,所以加了这么个参数。
pipi_dist_ang
pipi_dist_ang: to calculate the distance and angles between two rings or between one ring and one vector defined by atom group or command line.
:examples:
dit pipi_dist_ang -f test.gro -n test.ndx
dit pipi_dist_ang -f test.gro -n test.ndx -select ring1 ring2
dit pipi_dist_ang -f test.gro -n test.ndx -select ring1 -vg
dit pipi_dist_ang -f test.gro -n test.ndx -select ring1 -vec 6 6 6
:parameters:
-f, --input
the gro file which contains frames of molecule coordinates
-n, --index
the index file which contains atom index groups of rings or vector groups
-b
the frame number to start calculation, default=0
-dt
the frame interval, default=1
-o, --output
the output filename to save results
-vg
whether to get vector from index group, default=False
-vec
specify the vector by command line, eg. -vec 6 6 6
-select
select the groups from command line
pipi_dist_ang主要是用于计算两个环(5-7元环)的几何中心距离以及环法向的夹角。
用户通过在索引文件中新增原子组,并在其中定义相应的环的原子序号,就可以利用此命令计算环与环的距离和角度了。DIT会读入指定的原子索引,然后计算得到几何中心以及法向向量,之后得到距离和角度。
此命令还支持自定义一个向量,以计算特定环的法向与指定向量的角度变化。用户可以通过-vec参数在命令行中指定向量,也可以直接在索引文件中新建一个包含两个原子的组,两个原子的坐标即可形成一个向量。
hbond
hbond: A useful method to process hbond related files generated by `gmx hbond`,
you can get hbond occupancy figure and table infos from `dit hbond`.
note: the hbond names will show on figure only when number of hbonds
less than 10.
:examples:
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -c 5 7
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -c 5 7 0-3
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -o test.png -csv test.csv -ns
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -hnf d_resname(d_resnum)@d_atomname(d_atomnum)->h_atomname(h_atomnum)...a_resname(a_resnum)@a_atomname(a_atomnum)
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -hnf "d_resname@d_atomname -> h_atomname ... a_resname@a_atomname"
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -hnf "d_atomname@h_atomname -> a_atomname"
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -hnf "number"
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -hnf id -so AND0,2,5-7
dit hbond -f md.gro -n hbond.ndx -m hbmap.xpm -hnf id -so OR0,2,5-7
:parameters:
-f, --input
the gro file which contains one frame of molecule coordinates,
used to get atom names.
-n, --index
the index file generated by `gmx hbond`
-m, --map
the hbond map file (hbmap.xpm) generated by `gmx hbond`
-c, --select (optional)
select the hbond id to only show the selected hbonds
-o, --output (optional)
the figure name you want to save
-csv, --csv (optional)
save the table infos into a csv whose name is specified here
-ns, --noshow (optional)
whether not to show figure, useful on computer without gui
-hnf, --hbond_name_format (optional)
define the hbond name format by user!
Each atom has four features: resname, resnum, atomname, atomnum.
Distinguish donor, hydrogen, acceptor by adding one prefix to each
feature, like: d_resname, a_resnum, h_atomname. So you may able to
define hbond name style by:
'd_resname(d_resnum)@d_atomname(d_atomnum)->h_atomname(h_atomnum)...a_resname(a_resnum)@a_atomname(a_atomnum)'
which is the default style, or also you could specify
'd_atomname@h_atomname...a_atomname'
or some format you would like.
Or you could just set hnf to be 'number' or 'id' to show hbond id on figure
-genscript, --genscript (optional)
whether to generate scripts for calculating distance and angle of hbonds
)
-cda, --calc_distance_angle (optional)
whether to calculate distance and angle of hbonds from distance xvg file and angle xvg file
-distancefile, --distancefile (optional)
distance file of hbonds for input
-anglefile, --anglefile (optional)
angle file of hbonds for input
-xs, --xshrink (optional)
specify a factor for multiplication of x-axis. default == 1.0
For instance, if "-xs 0.001" is specified, all x-axis value of xvg
will multiply this value. x-axis 1000 will be shown as 1.
Useful for converting the unit (ps) of time into (ns). Don't forget
to change xlabel too after specifing -xs.
-x, --xlabel (optional)
specify the xlabel of figures
-so, --set_operation (optional)
use AND or OR to perform set operation on hbonds, eg. `AND1,2,5-6`
or `OR0,2-4,7`. The AND set or OR set of hbonds whose ids were after
key words will be calculated and shown in occupancy figure.
hbond命令主要用于生成氢键占有率图、给出氢键占有率信息。
gmx hbond命令执行结束之后会生成氢键的索引文件、氢键数量的xvg文件以及氢键占有率的xpm文件,而dit hbond命令需要读入一个体系的gro文件、氢键索引文件、氢键xpm文件。索引文件和gro文件主要用于获取形成氢键的原子的名称等信息,而xpm文件则包含了氢键占有率的信息。此命令会依赖这三个文件, 绘制出氢键占有率图,并在命令行中输出每个氢键的占有率。
用户可以通过-hnf参数自定义氢键的名称格式。每个原子包含四种属性:resname、resnum、atomname、atomnum,而前缀d_、a_、h_则分别代表了供体、受体、氢原子。用户将这些关键词组合在一起,即可定义自己需要的氢键名称格式。当然,也可以将-hnf设定为number,这将让DIT直接输出氢键的序号作为氢键的名字。
-genscript、-cda、-distancefile以及-anglefile是为了计算氢键的角度和距离而设置的。
使用步骤如下:
首先使用
gmx hbond命令得到氢键的xpm文件,hbond.ndx索引文件。此处一定记得加上-merge no这个参数,来避免同一供体上不同氢原子被合并使用
dit hbond绘制占有率图,给出占有率图表等信息,同时加上-genscript生成计算氢键距离和角度的索引文件以及控制脚本。命令可以为dit hbond -f test.gro -n hbond.ndx -m hbond.xpm -genscript,当然,还可以通过-c参数选择要计算的氢键。得到计算距离和角度的索引(hbdist.ndx和hbang.ndx)之后,通过运行生成的计算脚本(run_hbdist.sh和run_hbang.sh)调用gmx来生成相应的距离和角度的xvg文件。当然,这个计算脚本需要根据你的情况调整一下其中的文件名等内容。
通过命令
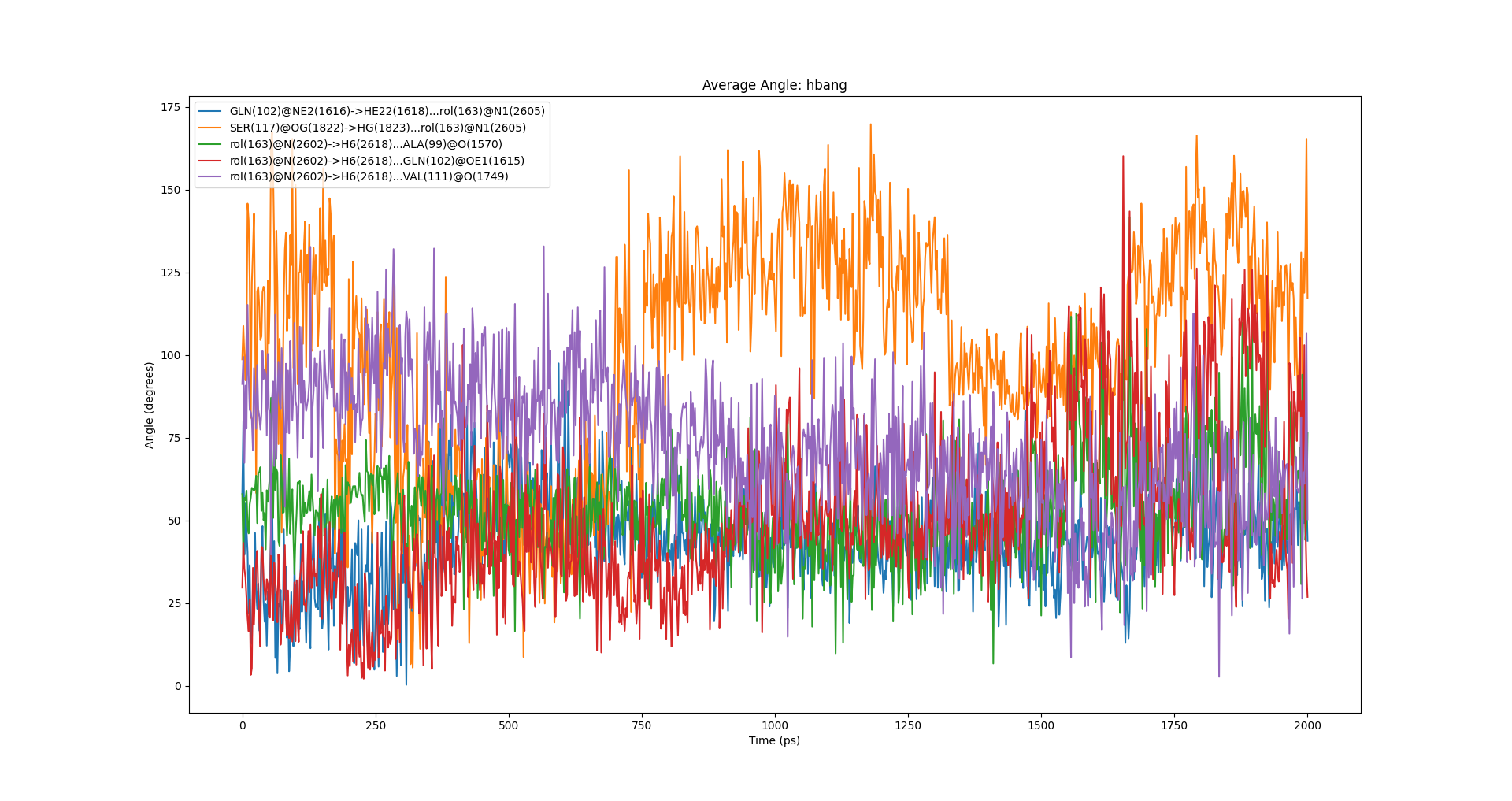
dit hbond -f test.gro -n hbond.ndx -m hbond.xpm -cda -distancefile hbdist.xvg -anglefile hbang.xvg -csv hhh.csv即可计算得到氢键距离和时间的折线图以及相应的统计表格,包含了氢键平均距离和平均角度。自然这个距离和角度的平均是在氢键形成的情况下统计的。最后的结果还可以通过-csv参数输出到文档。
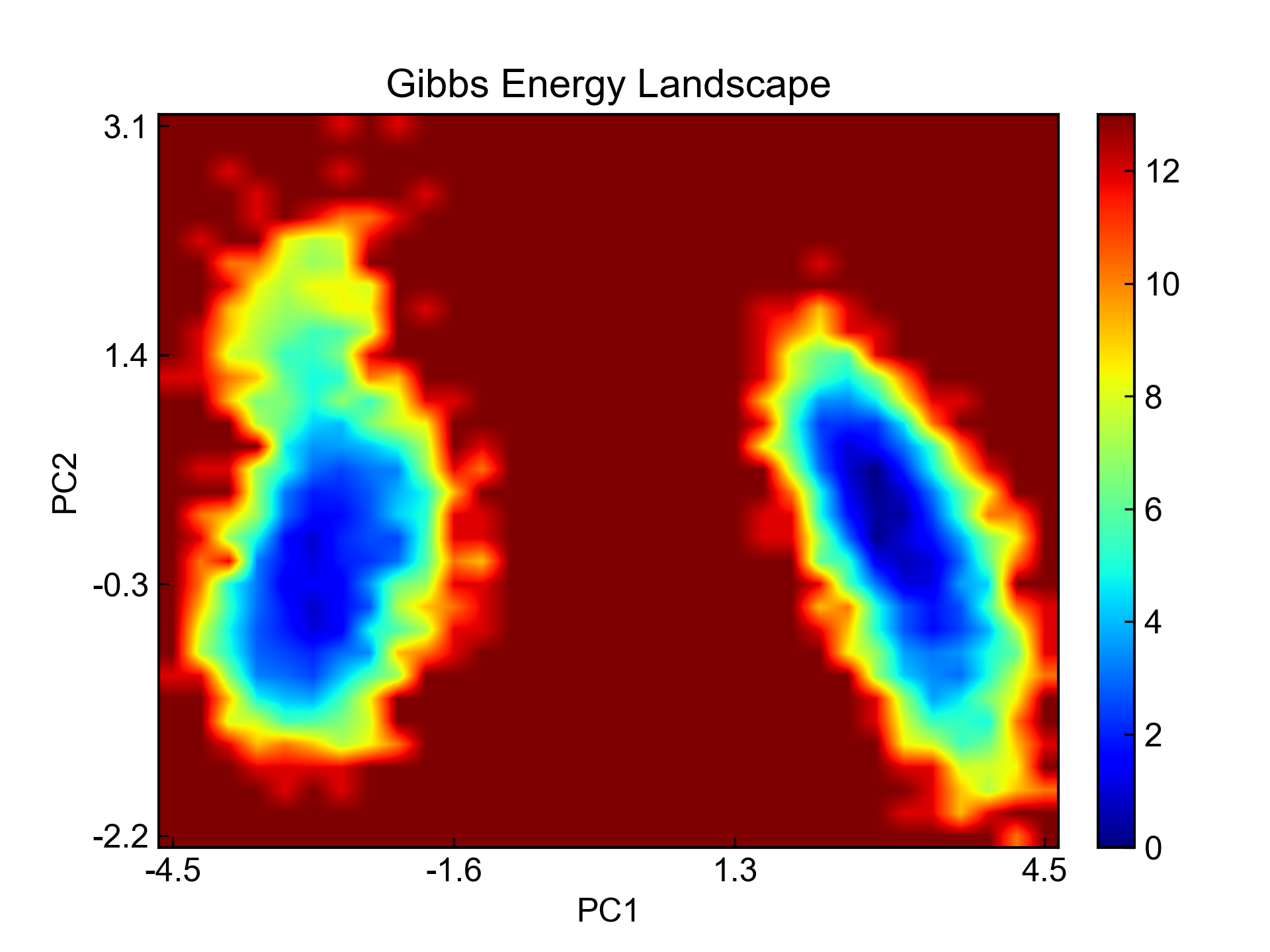
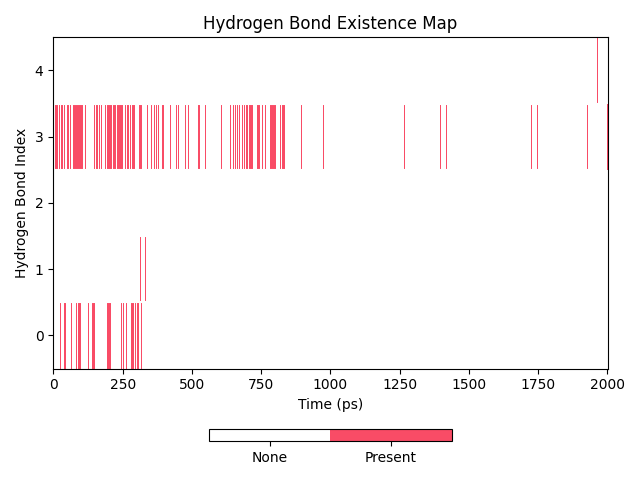
上述过程能够看到的图表有:
氢键占有率图:
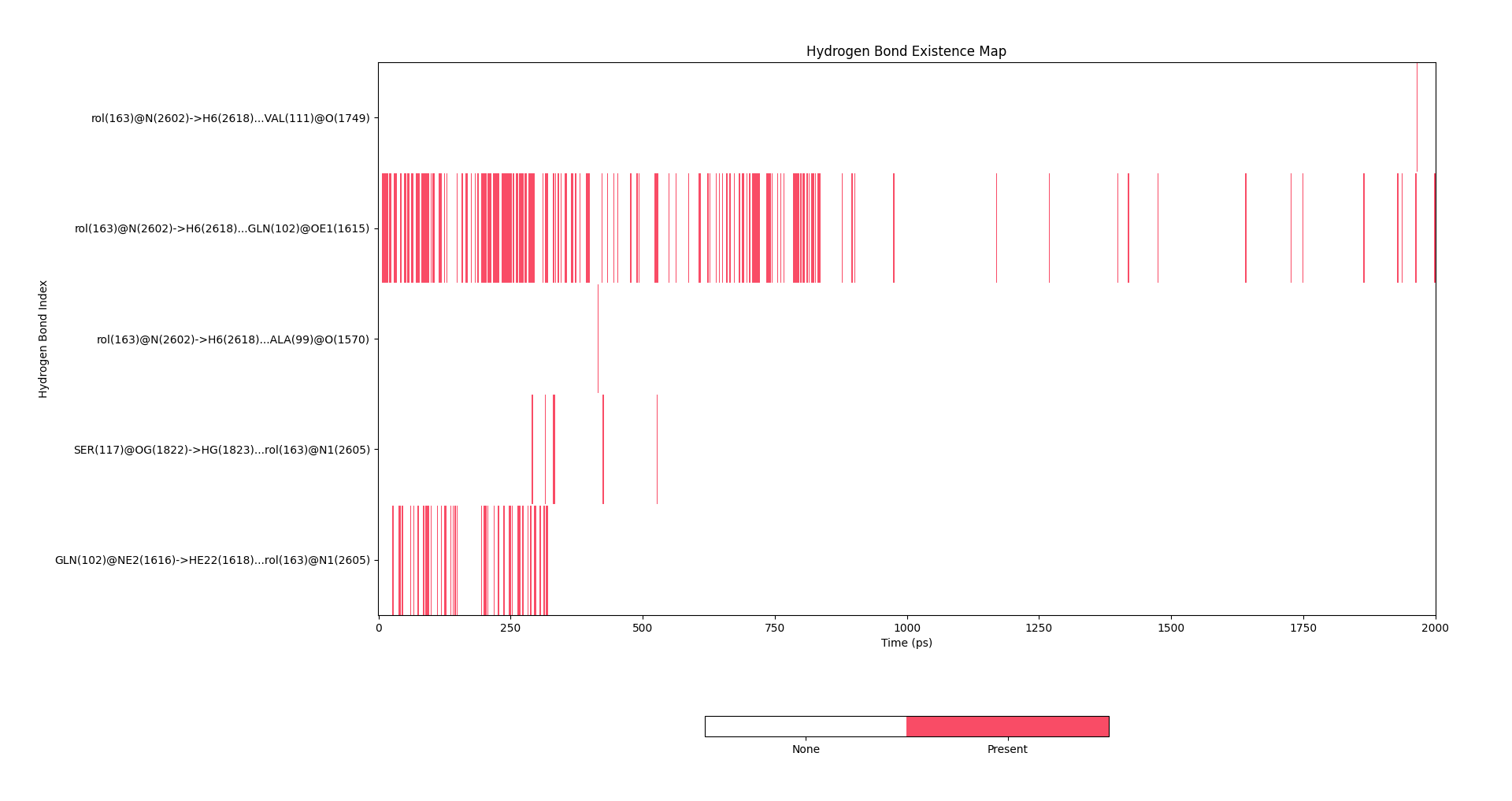
氢键距离随时间变化图:
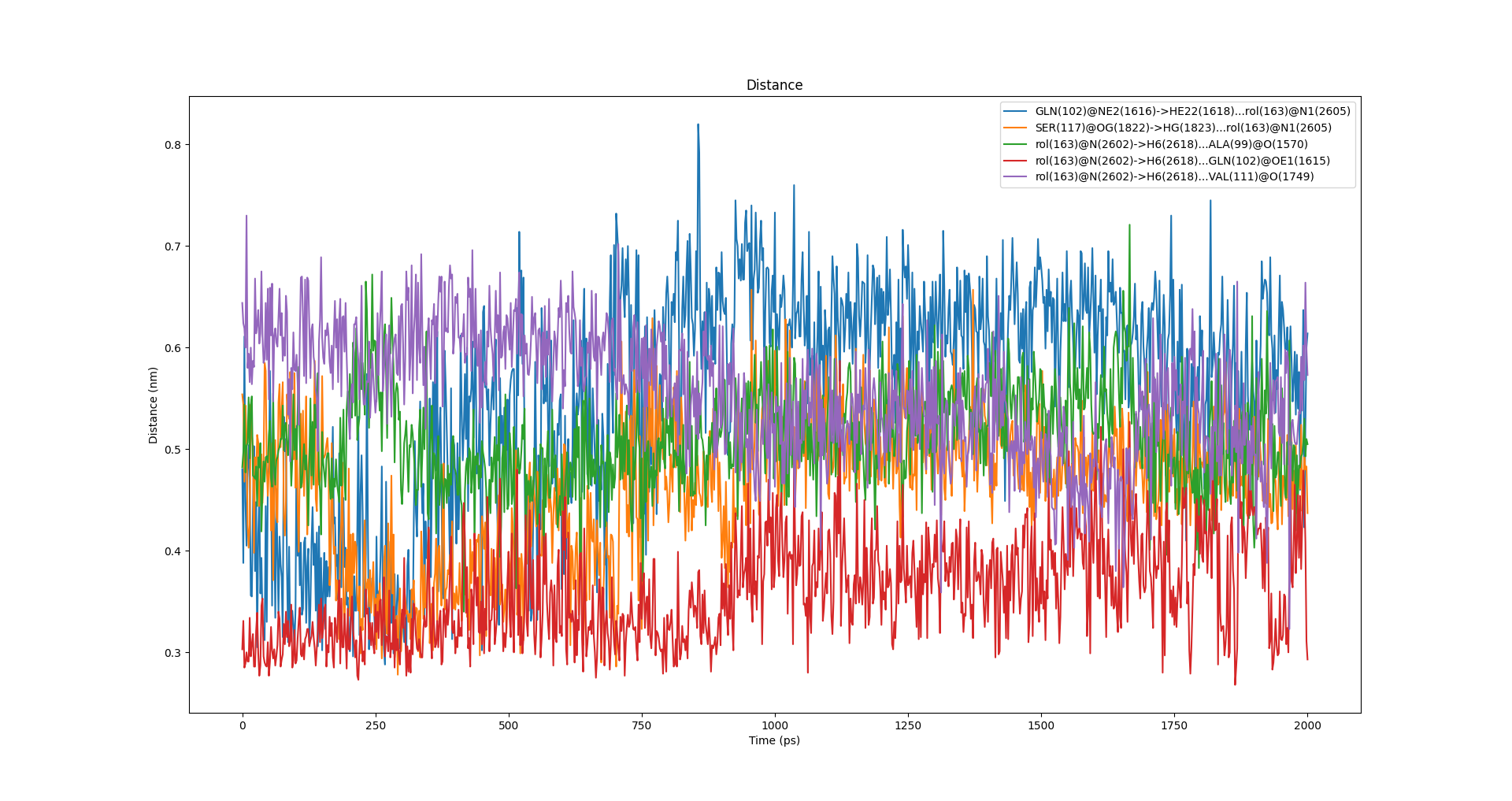
氢键角度随时间变化图:
得到的表格如下:
-------------------------------------------------------------------------------------------------------------------
id donor->hydrogen...acceptor occupancy(%) Present/Frames Distance (nm) Angle (°)
-------------------------------------------------------------------------------------------------------------------
0 GLN(102)@NE2(1616)->HE22(1618)...rol(163)@N1(2605) 4.60 46/1001 0.3219 ± 0.0167 17.91 ± 7.05
1 SER(117)@OG(1822)->HG(1823)...rol(163)@N1(2605) 0.60 6/1001 0.3102 ± 0.0218 12.75 ± 5.86
2 rol(163)@N(2602)->H6(2618)...ALA(99)@O(1570) 0.10 1/1001 0.3400 ± nan 23.03 ± nan
3 rol(163)@N(2602)->H6(2618)...GLN(102)@OE1(1615) 18.98 190/1001 0.3088 ± 0.0175 20.56 ± 7.38
4 rol(163)@N(2602)->H6(2618)...VAL(111)@O(1749) 0.10 1/1001 0.3230 ± nan 15.73 ± nan
-------------------------------------------------------------------------------------------------------------------
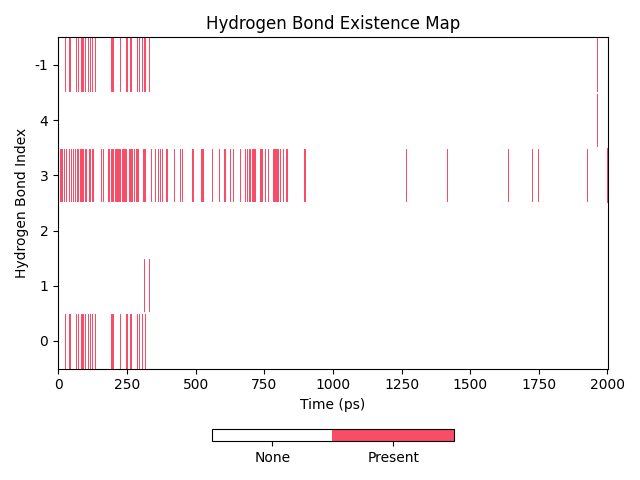
dit hbond还有一个功能,可以对氢键的占有率图进行操作,可以对氢键进行集合操作,例如求出多个氢键同时存在的占有率,或者多个氢键任一一个存在的占有率。这些操作通过-so(-set_operation)参数进行。此参数接受的参数格式为AND或者OR开头,后面接逗号分隔的氢键id,例如AND2,4-6,7或者OR0-4,6。这里的数字指代的是氢键id,可以通过-hnf id看到氢键的id。id从0开始计数,这里的AND2,4-6,7即为对第3、第5到第7(包括)和第8个氢键执行AND操作,得到的就是这些氢键同时存在的占有率。
下面是两个例子:
dit hbond -f test.gro -n hbond.ndx -m hbond.xpm -hnf id
dit hbond -f test.gro -n hbond.ndx -m hbond.xpm -hnf id -so AND0,3
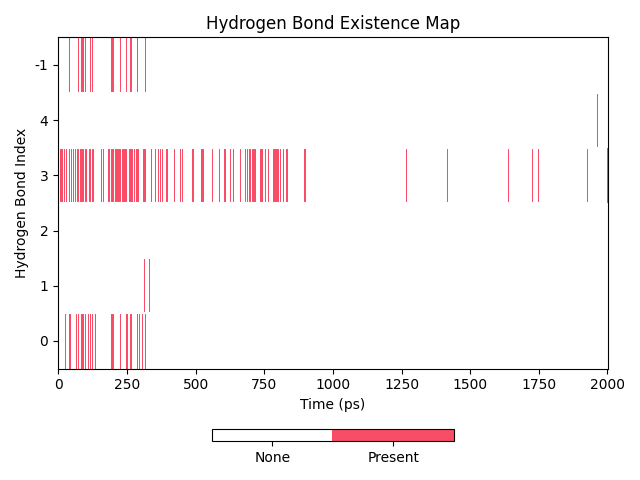
图上的-1指代的氢键即为集合操作之后的结果,如果没有设置-hnf id的话,这里会显示为AND0,3。同时表格也会给出相关的占有率信息。
dit hbond -f test.gro -n hbond.ndx -m hbond.xpm -hnf id -so OR0-2,4
dssp
dssp命令用于处理gmx do_dssp命令得到的xpm文件。共有3个小功能:
绘制此xpm文件。用户也可通过
dit xpm_show来可视化此文件绘制残基二级结构占比随时间的变化图。用户也可通过
gmx do_dssp命令得到的二级结构含量变化文件使用dit xvg_show_stack得到。同时会在当前文件夹生成residue_occupancy_data.csv绘制时间二级结构占比随残基的变化图。也即考察在整个分析采样过程中,某一个残基所形成的二级结构的时间占比。同时会在当前文件夹生成time_occupancy_data.csv
dssp: read in one xpm of secondary structure file, output the DSSP plot,
residue occupancy vs time figure, time occupancy vs residue figure.
:examples:
dit dssp -f dssp.xpm
dit dssp -f dssp.xpm -xs 0.001 -x Time(ns)
:parameters:
-f, --input
input xpm of secondary structure
-x, --xlabel (optional)
specify the xlabel of xpm
-y, --ylabel (optional)
specify the ylabel of xpm
-t, --title (optional)
specify the title of xpm
-xs, --xshrink (optional)
specify a factor for multiplication of x-axis. default == 1.0
For instance, if "-xs 0.001" is specified, all x-axis value of xvg
will multiply this value. x-axis 1000 will be shown as 1.
Useful for converting the unit (ps) of time into (ns). Don't forget
to change xlabel too after specifing -xs.
例如对于dssp.xpm,执行:
dit dssp -f dssp.xpm -xs 0.001 -x Time(ns)
首先得到xpm文件的可视化图:
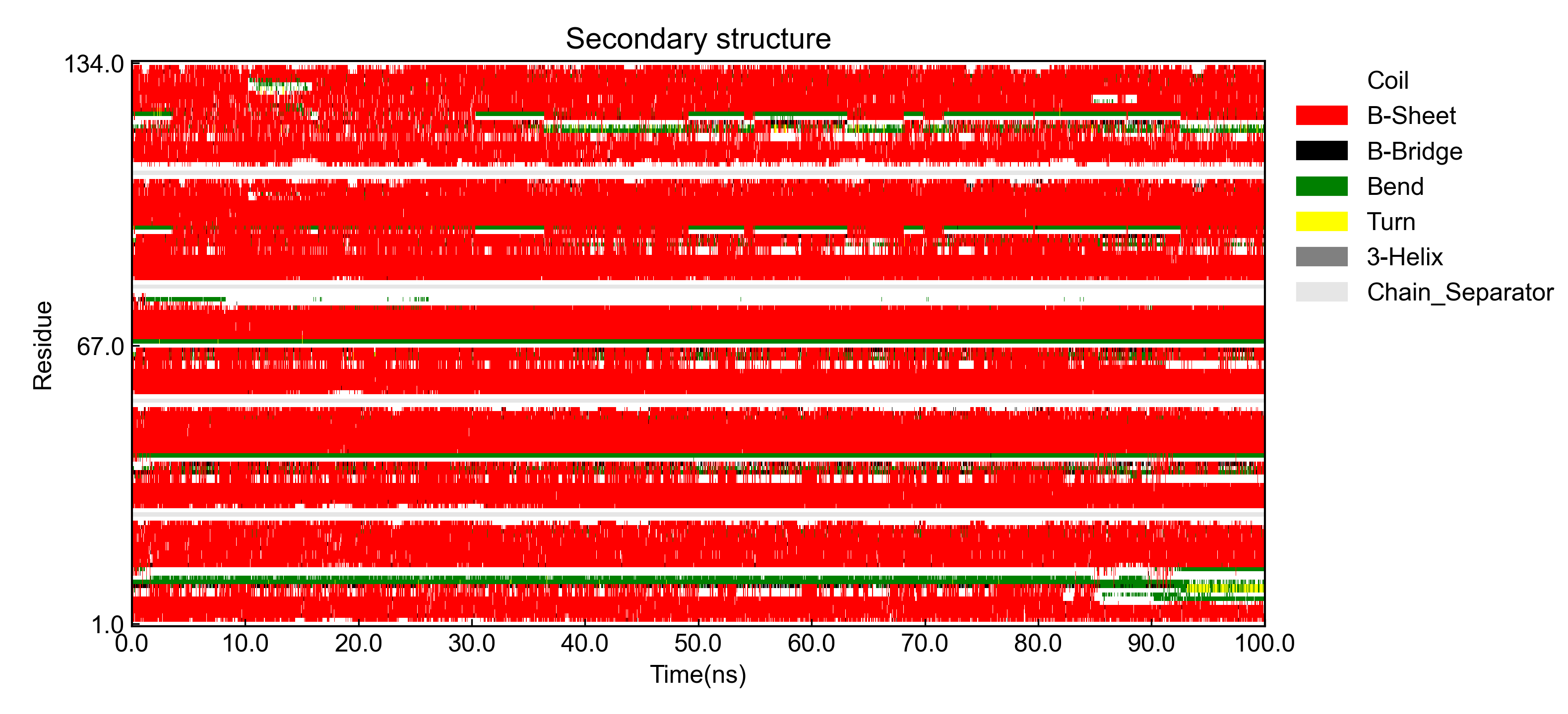
然后得到残基二级结构含量随时间的变化图:
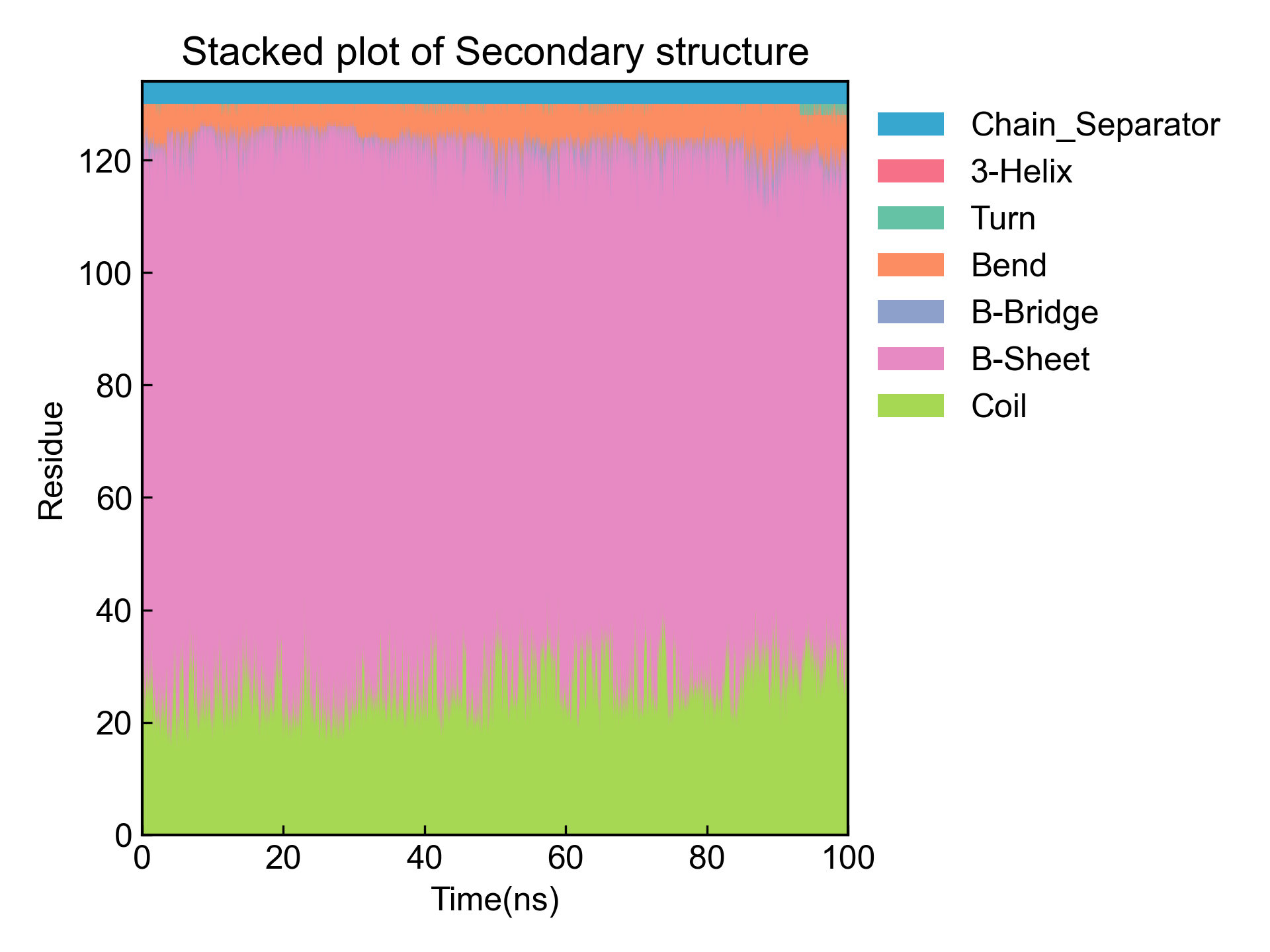
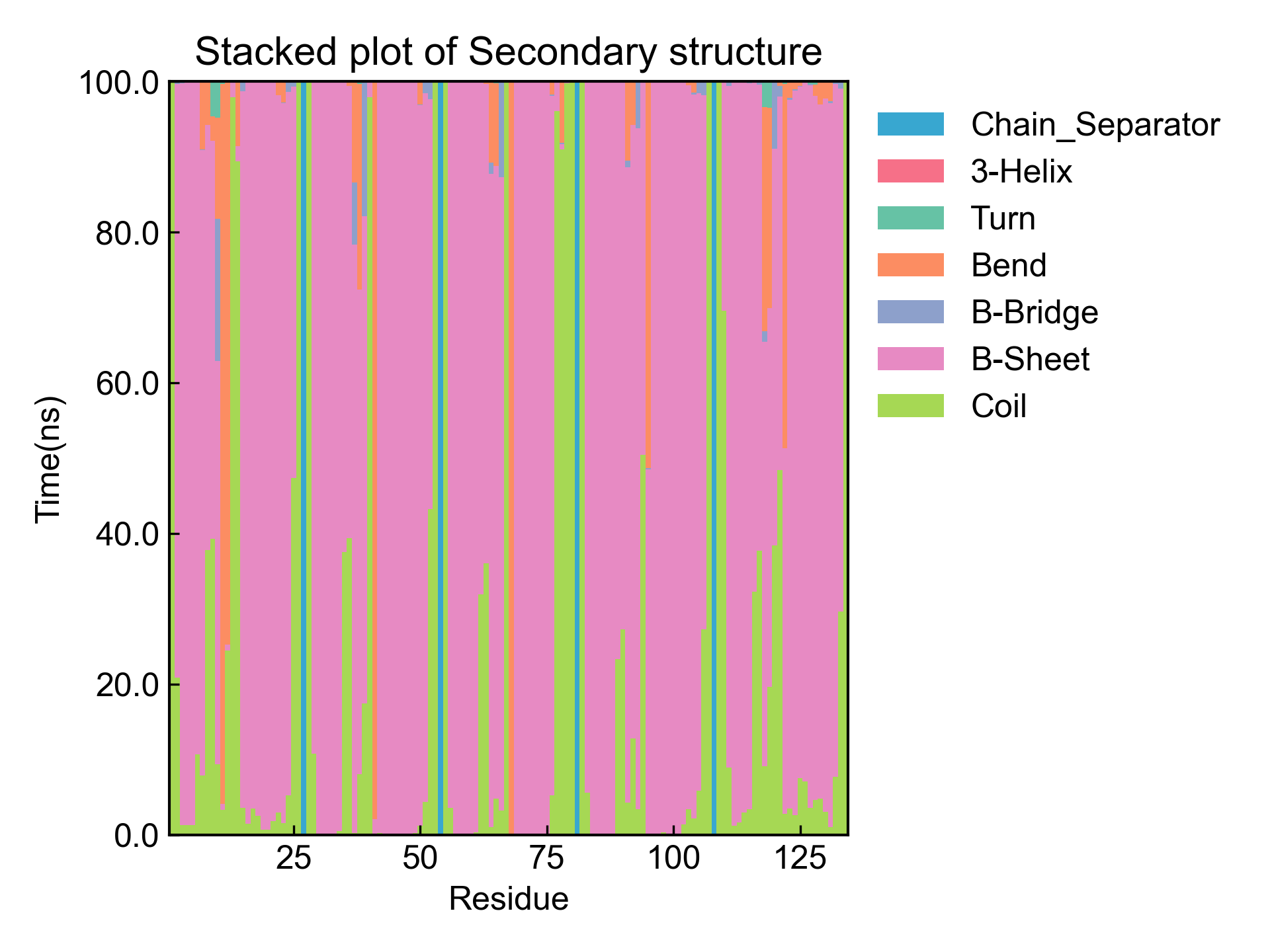
最后得到时间二级结构含量随残基的变化图:
mol_map
此命令不建议使用,算法上仍有一些问题需要解决。
如果两个分子构象完全相同,但是每个原子的坐标不同,可以通过此命令将两个分子的坐标替换,也即将分子从一个坐标转换到另一个坐标。
dccm_ascii
dccm_ascii: generate dynamic cross correlation matrix (DCCM) from dat file
generated by `gmx covar`.
:examples:
dit dccm_ascii -f covapic.dat
dit dccm_ascii -f covapic.dat -m gaussian -o res.png -noshow
dit dccm_ascii -f covapic.dat -x Res -y Res -t DCCM
:parameters:
-f, --input
the dat file generated by `gmx covar` which stored the covarience
matrix information
-o, --output
specify the output figure name
-m, --mode
specify the figure style, "origin", "gaussian", "bio3d"(default)
-ns, --noshow
whether not to show figure
-x, --xlabel
specify the xlabel of figure
-y, --ylabel
specify the ylabel of figure
-t, --title
specify the title of figure
dccm_ascii命令的主要功能是将gmx covar命令生成的协方差矩阵的dat文本文件转换成动态互相关矩阵。此命令读取协方差矩阵数据处理成DCCM数据之后绘图,可选的绘图样式有三种,origin、gaussian以及用matplotlib模仿的bio3d的绘图风格。
show_style
使用show_style在当前工作目录中生成DIT在用的绘图格式文件,可以通过修改该文件来控制绘图的相关格式,如字体字号等等。
future features
v0.4.8的版本发布之后,后续只会有bug修复的内容更新,不会再有新的功能。22年3月发布DIT以来,已经经历了几次小的版本迭代,笔者越来越感觉早期设计的逻辑框架不是很顺手。早期预期的功能没有这么多,因而核心的功能写得不够简洁明白,后面的一些东西也就不好加上去了,也无法确保程序的正确性。 因而,在v0.4.8之后,我会着手开发v0.5.0。这一版本会将DIT完全重构一遍,以下方面将会被优先考虑到:
各类文件的解析与数据转换将被独立出来
所有的命令行参数将被重新设计
增加更多绘图引擎,除了matplotlib之外,还有plotext和gunuplot等
增加对分析流程的支持
帮助信息和文档将更详细清晰
更好的命令行信息输出
更完善的单元测试
增加对残基接触矩阵的支持
……
Cite DuIvyTools
DuIvyTools目前是基于GPLv3协议开源的。我欢迎大家在日常工作中使用和修改,但不得以任何理由使用DuIvyTools牟利,包括但不限于付费获取、商业使用等等。
Cite DuIvyTools by:
![]()